


Ce dossier est associé à l’épisode #ps355 que vous pouvez réécouter ici.
J’espère ne va pas vous faire mal au crâne, puisqu’on va traiter d’une discipline… pas très limpide. Si l’astrophysique traite de planètes, la chimie d’agencement d’atomes et de molécules, les maths de chiffres et la biologie du vivant… il y a en réalité bien plus de sous domaines, qui sont des univers à eux seuls. Lors de chaque intro, Irène me présente comme une “biologiste moléculaire”…
Pour mieux vous introduire à ce qu’est la “biologie moléculaire”, j’aimerais qu’on se repenche rapidement sur les grandes expériences qui nous ont permis de comprendre comment ça fonctionne le vivant, et comment on en est arrivé à cette discipline un peu bâtarde, qui en mélange beaucoup d’autres. Il y a déjà un épisode de podcast science sur l’ADN (Episode 161 avec Vincent Judis) que je vous invite à écouter. J’ai essayé de ne pas être trop redondante avec cet épisode, mais j’aimerais quand même revenir un peu sur l’histoire de comment on en est arrivé là…
- Les grandes découvertes et le fondement de la biologie moléculaire.
Comme on est dans la “biologie”, vous vous doutez bien qu’on va rester sur des choses vivantes. Et comme on parle de “moléculaire” – vous vous doutez bien qu’on va parler de “molécules”, ou en tout cas de choses à une très petite échelle.
Alors, le premier pas vers ce monde microscopique, ça a été d’inventer… le microscope. Et concernant cet objet, la date d’invention ne fait pas consensus : Irène a fait un super dossier dans l’épisode 159 sur toute l’histoire de la microscopie (je vous invite à le lire) – mais en tout grâce à ces outils qui agrandissent ce que l’on peut voir, de nombreux naturalistes, scientifiques se sont mis à regarder le monde vivant avec un oeil nouveau (littéralement, et pas juste en métaphore). L’un des personnages les plus connu sera Anthoni van Loeewenhoeck, néerlandais ayant fait des grosses améliorations au microscope, qui lui ont permis de faire les premières observations détaillées de tout ce qui lui passait sous la main… y compris pour la première fois des petits micro-organismes indépendants. On appelle alors ces “animaux” microscopiques des “animalcules”.
Une célèbre image qu’on attribue à Robert Hooke en 1665 – une observation rapprochée de coupes d’écorce d’arbre – montre que la “peau” de l’arbre est constituée de petits carrés transparents, entourés de parois. C’est lui qui nommera ces structures “cellules” du mot “cellula” qui désignait les petites chambres des moines. Et le terme est très pertinent, puisque la cellule, c’est une unité vivante indépendante. La plus petite unité vivante capable de se reproduire toute seule.
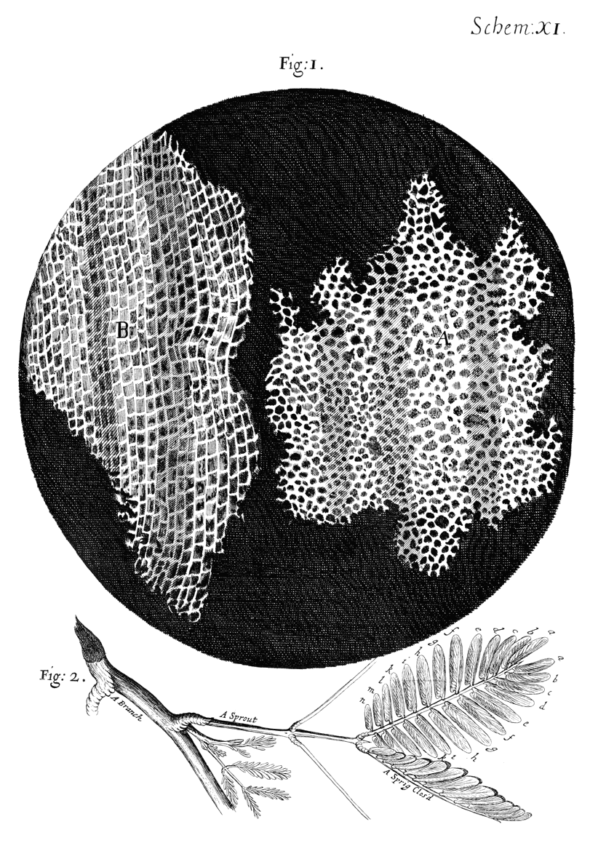
Bon, on est content… on sait que tous les organismes vivants sont constitués d’une ou plusieurs cellules. Et on commence à s’intéresser à ce qu’il y a dedans.
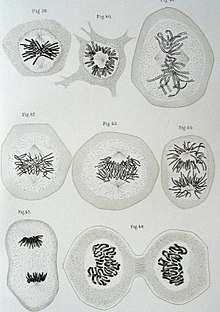
Très rapidement, les premiers cytologistes, ceux qui s’intéressent à la cellule se rendent compte que dans les cellules de beaucoup d’organismes pluricellulaires, il y a des noyaux, dans lesquels est contenu un truc qui se colore, la “chromatine”, fait d’un enchevêtrement de bâtonnets qui se colore eux aussi les “chromosomes” (corps colorés en latin). C’est à Walther Flemming – un biologiste allemand (et pas Alexandre) que l’on doit les premières descriptions détaillées du phénomène qu’on appelle encore aujourd’hui mitose en 1882 (du grec mitos= filament), et à Heinrich Willelhm Waldeyer que l’on doit le terme chromosome en 1889. (et neurone aussi, d’ailleurs)
Entre temps, Mendel, autrichien – avait cultivé ses fameux petits pois et déterminé les lois de l’hérédité aux alentours de 1865 – j’aime beaucoup cette histoire, mais vous la connaissez, je ne vais pas m’étendre. Du côté de l’Allemagne, deux chercheurs Oscar Hertwig et Eduard Strasburger (qui malgré son nom ne vivait pas à Strasbourg) – l’un biologiste animal, l’autre biologiste végétal s’intéresse tous les deux à l’embryologie. En 1876, Hertwig montre que la fécondation est l’union de deux noyaux d’un gamète mâle avec un gamète femelle. Strasburger fait la même chose sur des gamètes de plantes, et en 1880, il se disent que si les caractères doivent se transmettre d’une génération à l’autre… ce doit être à cause de quelque chose dans le noyau. En 1903, Walter Sutton , un américain, qui travaille sur la méiose chez la sauterelle et Theodor Boveri, un allemand qui travaille sur le développement embryonnaire des oursins proposent indépendamment que… peut être, les chromosomes pourraient porter l’information génétique.
Citons aussi Edmond Wilson, un américain. En observant la division des cellules spinales au stade pré-différencié (blastocyste) chez les mollusques, les vers plats et les annélides, il conclut que les mêmes organes viennent du même groupe de cellules et donc que tous ces organismes doivent nécessairement avoir un ancêtre commun. Il découvre également le rôle du chromosome XY dans la détermination du sexe en 1905. Nettie Stevens (1861-1912), une généticienne américaine, fait une découverte semblable la même année mais de façon indépendante. Wilhelm Johannsen, botaniste et généticien danois, suite à ses expériences de croisement sur des haricots propose le terme du “gène” et introduit les concepts de génotype (ce qui se transmet d’une génération à l’autre) et de phénotype (le résultat de ce qui s’est transmis sur l’apparence). Tout ça alors qu’on ne sait même pas ce qui est le support de l’information génétique !
Un des mentors de Nettie Stevens, Thomas Hunt Morgan, ignore et dément la théorie chromosomique de l’hérédité pendant des années, jusqu’à ce qu’il tombe fortuitement dans ses populations de mouche en élevage un mâle drosophile aux yeux blancs. Il note que la descendance d’un croisement de ce mâle aux yeux blancs avec une femelle aux yeux rouges suggère que le caractère « yeux blancs » est récessif par rapport au caractère yeux rouges. Morgan nomme « white » le gène gouvernant ce genre de caractère, inaugurant ainsi une tradition des généticiens de la drosophile qui consiste à nommer les gènes d’après le phénotype de leurs allèles mutants. Morgan remarque également que parmi les descendants d’un croisement de femelles mutantes aux yeux blancs avec des mâles sauvages aux yeux rouges, seuls les mâles présentent des yeux blancs. À partir de ce résultat, il conclut que (1) des traits phénotypiques (que l’on voit) sont liés au sexe, (2) que le trait « couleur de l’œil » est probablement porté par le chromosome sexuel, (3) que d’autres gènes sont probablement portés par d’autres chromosomes. C’est lui qui fait les premières cartes génétiques en utilisant le phénomène de recombinaison. AH. On avance. 1911. Il n’a pas eu de prix Nobel mais c’est lui qu’on a retenu pour cette théorie, alors qu’il a eu des prédécesseurs.
En 1941, George Beadle et Edward Tatum se rendent compte en isolant des mutants d’un champignon, Neurospora crassa – que certains mutants n’étaient plus capables de pousser sur un milieu de culture minimal. C’est à dire qu’il leur manque la capacité de produire eux mêmes certains métabolites/ certains nécéssaires pour pousser. Et eux savent que transformer les molécules dans une cellule, c’est le job de certaines protéines actives : les enzymes. En plus, en croisant les champignons et en faisant des analyses génétiques, ils deviennent convaincus que c’est lié à un seul gène. Ce sont les premiers à postuler qu’un gène, ce doit correspondre à une enzyme.
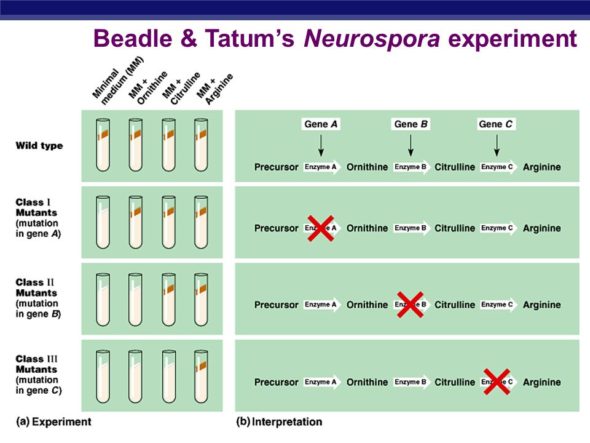
Là c’est intéressant, on est en 1941, on postule qu’un gène, ça doit correspondre à une enzyme… mais on ne sait toujours pas qu’est ce que c’est qu’un gène, si ce n’est que c’est vaguement dans les chromosomes, qu’il y a des acides nucléiques et des protéines, et que ça se transmet à la descendance.
Dans cet intervalle de temps, des gens un peu versés dans la chimie s’étaient eux aussi intéressés à ce que contenait une cellule. Gerardus Johannes Mulder (pas encore accompagné de Scully), un chimiste néerlandais en travaillant sur le blanc d’oeuf se rend compte qu’il est composé de substances organiques. Selon lui, ces substances composeraient de la même façon tous les organismes et c’est le premier à avancer que les animaux tireraient leurs protéines des plantes… il les baptise “protéines” – du grec proteos (premier). Ca c’est en 1835.
Un peu plus tard, un certain Friedrich Miescher (suisse) découvre la composition des globules blancs. En lavant des bandages couverts de pus dans un hôpital grâce à une solution saline, puis en y versant une solution saline, il se rend compte que les noyaux des globules précipitent. Et que ces précipités sont constitués de molécules contenant des atomes de phosphore… il l’observe aussi dans d’autres types de cellules, des oeufs de poule, du sperme de saumon… un peu partout ! Il baptise cette substance la “nucléine” en 1879.

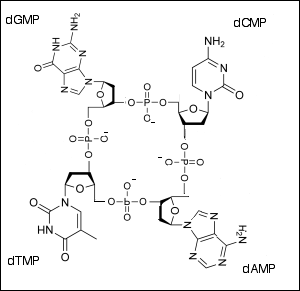
Et puis il y a Phoebus Levene, un grand biochimiste américain qui avant 1910 réussit à élucider précisément la nature chimique de la nucléine : l’acide désoxyribonucléique, 4 briques chimiques qui peuvent s’attacher les unes aux autres. Il se plante cependant en affirmant que l’ADN est constitué d’un enchaînement de tétranucléotides. Comme on sait alors que les protéines sont constituées de 20 briques chimiques diférentes, alors que les acides nucléiques sont constitués de 4 briques différentes – les possibilités pour “porter une information” sont plus nombreuses avec les protéines.
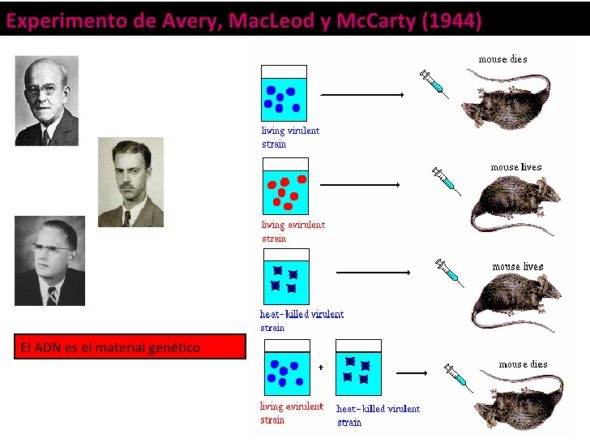
En 1928, les expériences de Frederick Griffith montrent qu’une souche de bactéries virulente que l’on détruit est capable de donner ses caractéristiques à une souche non virulente et postule l’existence d’un “principe transformant”. En 1944, Oswald Avery, MacLeod et McCarty en reprenant ces expériences et les poussent plus loin : cette fois ci, il incube les bactéries non virulentes – avec différents extraits des bactéries virulentes. Ils incubent donc soit avec des protéines, soit avec des acides nucléiques et constatent que la virulence n’est acquise que lorsqu’on injecte de l’ADN de bactérie virulente… donc que l’ADN doit être le support de ce caractère “virulent”.
Dans les annnée 1952, les expériences de Alfred Hershey et Martha Chase lèvent définitivement le doute : eux s’intéressent aux phages, donc aux virus qui infectent spécifiquement des bactéries. En marquant radioactivement soit l’ADN (en y incorporant du phosphate P32), soit les protéines en y incorporant du souffre – ils constatent que la radioactivité pénètre dans les bactéries seulement lorsqu’elle est incorporée à l’ADN, et que la radioactivité des protéines de l’enveloppe du virus ne se propage pas dans les bactéries. Les virus infectent donc bien en faisant rentrer de l’ADN dans les cellules. (devinez lequel des deux a eu le prix nobel).
On arrive en 1953 avec la structure en double hélice de l’ADN. Watson et Crick, ceux qu’on a retenu pour cet exploit disposaient déjà de pas mal d’indices : (i) la composition chimique de l’ADN (désoxyribose, bases azotées, et groupements phosphate) grâce à Levene ; (ii) les clichés de diffraction aux rayons X d’ADN cristallisé, clichés dus principalement à Rosalind Franklin et Maurice Wilkins. Ces clichés montrent une figure en croix, caractéristique des structures en hélice ;
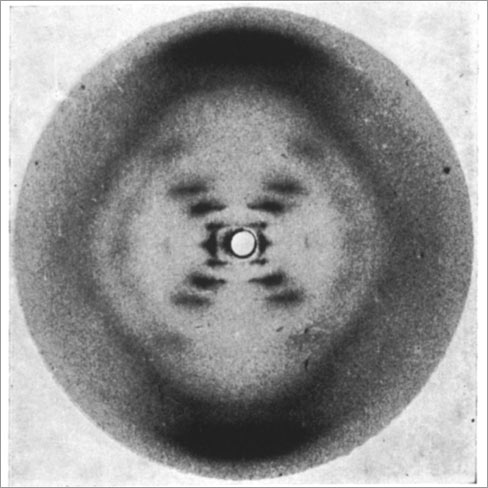
(iii) les travaux de Erwin Chargaff, qui avaient montré que pour toute molécule d’ADN, le nombre de molécules d’adénine est égal au nombre de molécules de thymine, et que celui de cytosine est égal à celui de guanine ; (iv) les analyses en microscopie électronique, qui avaient montré que le diamètre de la molécule d’ADN est de 20 Å, ce qui suggérait que cette molécule comportait deux chaînes de désoxyribose-phosphate. Du coup les gars ont fait des maquettes – et le seul modèle qui collait avec toutes ces données, c’est celui qu’on retient aujourd’hui. Deux brins constitués des groupements phosphates et des sucres forment une double hélice où les orientations de chacun des brins sont opposées. Sur les sucres de chacun des deux brins sont liées les bases azotées, chaque base d’un brin étant maintenue en vis-à-vis d’une base de l’autre brin par des liaisons hydrogène. Une cytosine fait toujours face à une guanine, et une adénine à une thymine. Les deux brins d’une molécule d’ADN sont dits complémentaires.

Et c’est seulement il y a 5 ans qu’on a clairement vu la structure en double hélice de l’ADN de nos propres yeux (et encore), avec le microscope à force atomatique (ATM)

Pourquoi je vous re-raconte tout ça ? Hé bien simplement pour vous rendre compte que souvent, on raconte l’histoire des sciences de façon… très linéaire. On dit souvent qu’un tel ou qu’un tel est le père d’une découverte. Et on oublie les autres. Tous les autres. La découverte de la structure de l’ADN n’est pas un historique linéaire, mais une multitude de timeline parallèles, dont les individus se croisent, échangent des idées, des découvertes… et… voilà, c’est comme ça que devrait fonctionner la recherche.
D’autre part, là on arrive à un truc intéressant, parce qu’on constate que un :
- On a des datas chimiques
- On a des datas d’observation
Mais on ne sait toujours pas comment on passe de l’un à l’autre. On sait que l’ADN est le support de l’information génétique, on connaît sa structure moléculaire mais on ne sait toujours pas comment cette information est portée, codée… Je vous renvoie à ce cours sur ce que je viens de vous expliquer.
Et c’est là que tout s’accélère et que tout devient encore plus intéressant.
En 1940 grâce au biologiste belge Jean Brachet qu’il existe deux molécules distinctes dans cette nucléine. Celle qu’on appelle l’acide thymonucléique est un composant des chromosomes et qu’il est synthétisé lorsque les cellules se divisent après la fécondation. Il met aussi en évidence l’existence d’acides zymonucléiques (ARN) dans tous les types cellulaires: dans le noyau, le nucléole et le cytoplasme de toutes les cellules (alors que l’on pensait à l’époque que ces molécules étaient caractéristiques des cellules végétales et des eucaryotes inférieurs tels que les levures).
Les études biochimiques sortent bien vite qu’il existe plusieurs types d’ARN dans les cellules – des longs, messagers qui correspondent à une copie de la séquence ADN – et que c’est cet ARN qui exporté dans le cytoplasme et utilisé pour faire des protéines. Ce sont les expériences de Brachet, dans lesquels il fait en sorte de marquer spécifiquement les ARN au tritium (un isotope radioactif de l’hydrogène) pendant un laps de temps court, pour le suivre dans la cellule. Il remarque que l’ARN est produit dans le noyau, et qu’ensuite il se déplace vers le cytoplasme. 1951.
Certains sont des ARN ribosomiques, qui conjointement à des protéines forment le ribosome, découvert en 1950 par George Emile Palade – grâce à la microscopie électronique et qui est nécessaire à la synthèse des protéines.
Et en 1954 Hoagland et Zamecnick découvrent une autre molécule, qui fait l’intermédiaire entre l’ARN messager et le ribosome : l’ARN de transfert. Pour une histoire de sa découverte, consultez ce lien. Ce sont littéralement des adaptateurs, des petits acides nucléiques de 60 à 100 nucléotides qui se plient en formes de petites croix, et dont un bout reconnaît l’ARN messager, et l’autre bout porte un acide aminé qui formera la protéine.
![]()
Ils parviennent aussi à générer les premiers systèmes de synthèse protéique “in vitro” en purifiant des ribosomes depuis des bactéries. C’est la porte ouverte pour craquer le code et ce sont deux chercheurs, Matthei et Nirenberg en 1957 qui s’y collent. L’expérience m’avait littéralement impressionnée à l’époque quand j’étais étudiante. (Je ne sais pas si je dois vous l’expliquer ou pas)
Après leurs travaux, on sait que sur l’ADN, un triplet d’acides nucléiques “le codon” correspond à un acide aminé, que certains codons signalent le début d’une protéine, et que d’autres en signalent la fin. Et donc nous sommes dans les années 60 et on en est au principe de base que l’on a retenu aujourd’hui et qu’on enseigne à l’école. Et je vais m’arrêter entre chaque point pour que vous puissiez poser vos questions.
- L’information génétique est portée par la molécule d’ADN, qui est un enchaînement d’acides nucléiques, qui forment une double hélice de deux brins dont les bases sont complémentaires.
- Cette information doit être répliquée (lors d’une étape de réplication) pour être passée à la descendance- la cellule fille, lors de la division cellulaire.
- Cette information est stockée dans le noyau et doit être copiée en molécule d’ARN messager, simple brin (lors du processus de transcription) pour être exportée dans un autre compartiment de la cellule, le cytoplasme.
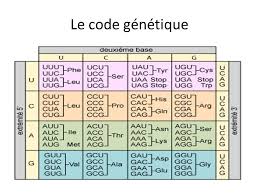
- La molécule d’ARN sera lue selon des règles précises lors d’une étape de traduction, pendant laquelle une machine moléculaire (le ribosome) fera correspondre chaque triplet de nucléotides à un acide aminé, avec une correspondance qu’on a répertorié : le “code génétique”.
- La chaîne d’acides aminés générées ont des propriétés physiques et chimiques qui leur donne des caractéristiques et des fonctions propres. Ce sont les protéines, qu’on appelle aussi des enzymes si elles ont une activité qui leur permet de transformer une molécule en un autre, donc de catalyser une réaction chimique. (D’ailleurs en réalité il y a 22 acides aminés différents dans les protéines et pas 20 – la pyrrolysine chez les archées, et la sélénocystéine dans certaines protéines chez les eucaryotes)
- Chaque protéine a une fonction (structurale, enzymatique, de signalisation, motrices, de défense, de stockage….) et l’ensemble de nos protéines constitue nos cellules. La boucle est bouclée.
Ca a l’air très abstrait comme ça, mais dans les notes de l’émission je vous laisse les liens de vidéos qui montrent la réplication, la transcription et la traduction modélisée.
Ces concepts fondamentaux, c’est ce qu’on appelle le “dogme” de la biologie moléculaire. Le socle de connaissance sur lequel repose tout notre travail quand on est biologiste moléculaire.
- Les grandes techniques de la biologie moléculaire.
Le terme “biologie moléculaire” est proposé en 1938 par Warren Weaver… qui n’était pas du tout biologiste (je ne sais pas trop le contexte mais bon, il a dit ça un jour dans sa vie). Mais du coup vous voyez que ce mot est antérieur aux fondements sus-mentionnés. La biologie moléculaire n’est pas en elle-même une discipline, c’est une expression commode pour désigner la « molécularisation » de la biologie. Sa transposition au monde de l’infiniment petit. Par contre, elle repose sur une série de méthodes et de stratégies, qui permettent d’accéder aux phénomènes moléculaires du vivant, parce qu’évidemment… on ne voit pas ce qui se passe. Et ces méthodes évoluent constamment, avec les progrès dans tous les domaines des sciences.
Purifier/Isoler des macromolécules
Alors je vais vous donner quelques méthodes d’analyse basiques de toutes ces molécules – mais déjà gardez en tête que pour analyser de l’ADN, de l’ARN, de la protéine… chaque type de molécule (et en l’occurrence de macro-molécule) à des propriétés physiques et chimiques différentes. Donc si vous voulez récupérer uniquement l’une des trois, on a des méthodes pour les extraire individuellement. Pour l’extraction d’ADN par exemple, vous pouvez assez simplement le faire à la maison : Prenez des cellules qui ont plein d’ADN – des cellules de banane par exemple – votre salive – de l’oignon… En gros l’ADN est dans le noyau, le noyau est dans la cellule. Donc vous pétez la membrane de la cellule (qui est faite d’une enveloppe de gras) avec du savon (comme vous faites la vaisselle, vous savez que le savon fait partir le gras). Savon qui va en plus dénaturer les protéines (donc leur faire perdre leur conformation) – vous rajoutez de l’eau et du sel pour dissoudre l’ADN, vous rajoutez de l’alcool ménager et pouf, l’ADN précipite en une jolie méduse blanche.
On fait pareil en laboratoire, mais en plus propre puisqu’on peut séparer les protéines de l’ADN en utilisant du phénol chloroforme. Les protéines vont rester dans la phase organique de solvant, alors que l’ADN va passer dans la phase aqueuse.
Donc je ne vais pas rentrer dans les détails plus que ça, mais partez de ce postulat : à partir de matériel vivant de notre choix, on peut récupérer le type de molécule qu’on veut.
Amplifier de l’ADN spécifiquement. L’histoire de la PCR.
Comme une cellule c’est très petit, souvent on travaille avec une petite quantité de matériel… et puis extraire tout l’ADN d’une cellule… ça fait beaucoup de matériel, beaucoup de gènes, beaucoup de trucs. Imaginez si on pouvait produire une grande quantité de juste la partie d’un ADN qui nous intéresse ! L’invention arrive vers 1986, grâce à Kary Mullis. Une petite parenthèse sur le personnage : il a écrit un livre qui s’appelle “Dancing naked in the mind field” où il raconterait son usage récréationnel du LSD, et sa rencontre avec un raton laveur luminescent qui lui aurait dit “Bonjour Docteur”. J’ai pas encore pris le temps de le lire… mais ça me donne envie.
Mais revenons sur la PCR : Quelques années plus tôt, on avait découvert les ADN polymérases – les protéines qui copient l’ADN. Et Karry Mullis a eu la bonne idée d’exploiter leurs capacités pour amplifier de l’ADN.
L’ADN est un double brin complémentaire. Dans la cellule ce double brin est ouvert par des enzymes et l’ADN pol le copie, en utilisant la complémentarité des bases. En face d’un A, on met un T, en face d’un C on met un G. Et pour commencer sa copie d’un brin simple, elle a simplement besoin d’un petit bout de copie pour commencer, ce qu’on appelle une “amorce” – double brin. Ducoup, si on fait en sorte de séparer les brins d’ADN en les chauffant, qu’on rajoute des amorces complémentaires de la zone qu’on veut amplifier, qu’on rajoute une Polymérase, des acides nucléiques libres et qu’on la laisse faire sa vie… elle nous copie notre ADN ! Top ! On passe d’un brin d’ADN à deux brins au premier cycle, puis de 2 à 4, puis de 4 à 16… et l’amplification est exponentielle !
Seulement chauffer l’ADN implique de dénaturer les protéines. Du coup à chaque cycle d’amplification, il faut remettre de l’enzyme fonctionnelle. Une des grosses améliorations arrive 2 ans plus tard dans un papier de Saiki et al. publié dans science en 1988, avec la découverte d’une ADN polymérase de bactérie thermophile : Thermus aquaticus. La bactérie vit dans des sources chaudes jusqu’à 80°C… et ducoup ses protéines sont thermostables et résistent à des fortes températures. Du coup à chaque cycle de PCR, plus besoin d’en remettre. On peut se contenter de mettre les tubes avec tout le matériel nécessaire dans une machine qui change de température : un thermocycleur, et c’est parti. On chauffe, ce qui dénature l’ADN. On refroidit un peu pour permettre aux amorces de se réhybrider (réattacher) à la zone d’ADN complémentaire, puis on se met à la température idéale de fonctionnement de l’enzyme, 72°C, qui va permettre de copier la molécule d’ADN.
Et PAF. Ca fait des chocapics. C’est encore aujourd’hui cette méthode qu’on utilise pour amplifier de l’ADN… en recherche, et aussi dans NCIS et les experts. C’est aussi une super méthode pour changer légèrement la séquence d’un fragment d’ADN et d’y introduire des mutations. Ou y rajouter des extrémités d’une séquence définie qui nous permet de faire du bricolage avec… je vais vous expliquer comment tout à l’heure.
Séparer les molécules en fonction de leur taille : électrophorèse.
Concrètement une fois que vous avez isolé des molécules – vous pouvez vouloir les trier. En fonction de leur taille par exemple. Une méthode très simple pour faire ça, c’est savoir que les acides nucléiques sont composés de phosphate. Le phosphate c’est un groupement qui a une charge négative – et ducoup qui va migrer vers la borne positive dans un courant électrique. Si on fait migrer l’ADN dans un maillage de gel (de l’agar), on sait que les petits fragments vont migrer plus vite et plus loin, tandis que les gros bouts d’ADN vont se traîner. Pour être sûr de la taille, on fait toujours migrer un mélange de bouts d’ADN de taille définie, qu’on appelle une “échelle de taille” ou “ladder”. Problème : mettez de l’ADN dans un gel et essayez de le voir. L’ADN n’a pas de couleur malheureusement. Pour révéler on utilise souvent une molécule fluorescente, le bromure d’éthidium (BET) qui s’intercale entre les bases ADN et émet une fluorescence orange lorsqu’on l’illumine aux ultraviolets.

Grâce à ces deux techniques, on arrive à développer la technique du séquençage.
A la fin des années 1970, deux scientifiques Walter Gilbert et Frédérik Sanger ont indépendamment une idée de génie pour accéder à la séquence de l’ADN. Ce qui leur a valu un prix nobel à tous les deux. Je vais vous parler de celle de Sanger : et si on modifiait chaque acides nucléiques (A,T,C,G) chimiquement pour que la polymérase soit obligée de s’arrêter pendant sa copie ?
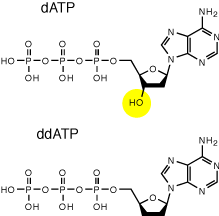
On obtiendrait statistiquement tout un tas de fragments d’ADN de différentes tailles, qui s’arrêtaient à chaque fois lorsqu’un de ces acide nucléique est incorporé.
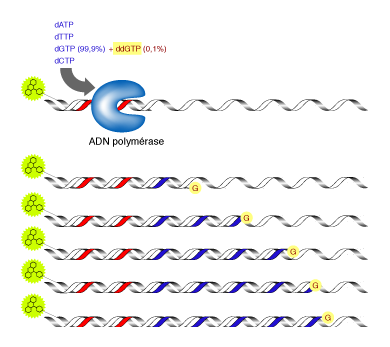
Ducoup il suffit de mettre le même bout d’ADN dans quatre tubes différents, la polymérase, les acides nucléiques dont un est modifié (soit le A, soit le T, soit le C, soit le G) – de faire migrer ça sur un gel… et de reconstituer la séquence du brin d’ADN en lisant sur le gel. En lisant du fragment le plus petit, au fragment le plus grand – donc du bas du gel vers le haut du gel si vous avez bien suivi mon explication sur l’électrophorèse… vous pouvez lire l’ADN !
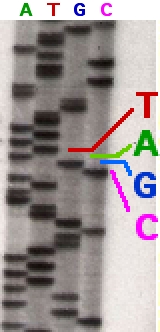
Une amélioration immédiate a été de transformer chaque nucléotide chimiquement pour les coupler à des signaux de fluorescence, ou faire en sorte qu’ils fassent de la lumière lorsqu’ils sont incorporés. Autrement dit, lors de la copie, chaque fois qu’un A va être incorporé, on a un pic lumineux d’une, chaque fois que c’est un T, un autre, etc… Et donc au fil de la lecture, on a des signaux distincts qui nous permettent d’avoir accès à la séquence. On récupère donc des fichiers qu’on appelle des “profils de séquençage”.
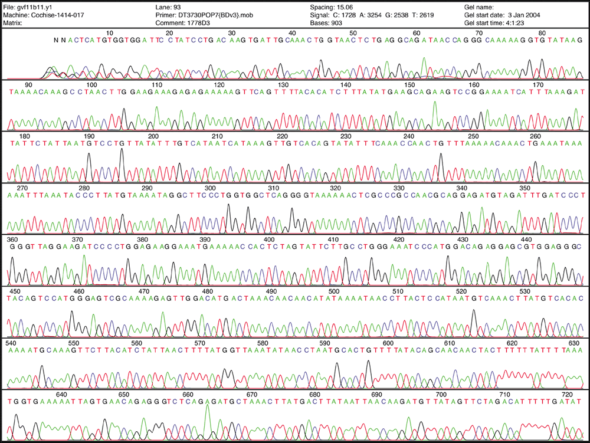
Pour vous donner une idée de la vitesse d’amélioration de cette méthode… alors qu’avant on pouvait passer une thèse entière à déterminer la séquence d’un gène manuellement, petit bout par petit bout, gel après gel… maintenant j’envoie 20 séquences à analyser dans une boîte aux lettres. Mes séquences partent dans une entreprise qui les séquence pendant la nuit, et le matin j’ai le résultat de mes séquences dans ma boîte mail.
Toujours grâce à cette séparation sur gel :
Ce qui est pratique, c’est qu’une fois qu’on a amplifié ce qui nous intéresse on peut le purifier, l’extraire du gel. Imaginez qu’on a amplifié le gène qui est responsable de la production d’insuline. Et que dans nos amorces on ait rajouté des séquences qui sont reconnues spécifiquement par des enzymes. On appelle ça des “sites de restriction” et ces sites peuvent être coupés par des enzymes qu’on appelle les enzymes de “restriction”. Elles ont été découvertes vers les années 60 par Werner Arber et son étudiante Daisy Dussoix… encore un prix Nobel, devinez lequel des deux ne l’a pas eu.
Maintenant imaginez qu’on ait un bout d’ADN artificiel circulaire qui ait la capacité de se répliquer tout seul dans un micro-organisme facile, une bactérie mettons. Imaginons que je coupe ce cercle d’ADN et que j’y colle mon gène. Ensuite imaginons que je mette ce bout d’ADN dans une bactérie qui va se l’approprier…
Mesdames et messieurs, nous venons de faire ce qu’un biologiste moléculaire un “clonage”. Prendre un bout d’ADN, l’insérer dans un ADN “véhicule” qu’on appelle un “vecteur” – et insérer ce vecteur dans un nouvel organisme pour qu’il l’amplifie et qu’il exprime le bout d’ADN en question.
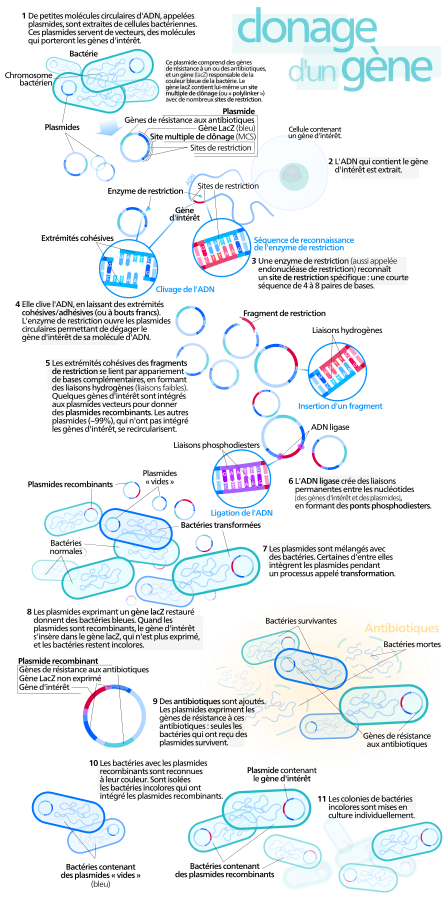
C’est grâce à cette méthode qu’on fait produire de l’insuline à des bactéries dans des bioréacteurs. Alors qu’avant on devait tuer et saigner des vaches pour purifier l’insuline à partir de leur sang.
Alors justement, l’insuline, c’est une petite protéine qui agit comme une hormone, et c’est Frédérick Sanger qui a découvert sa structure… il a d’ailleurs eu un deuxième Prix Nobel pour ça. Oui pourquoi se contenter d’un seul prix nobel ?
Du coup, pour séparer des protéines – on fait la même chose pour séparer des acides nucléiques, avec quelques adaptations techniques.
Séparer les protéines en fonction de leur taille : SDS-PAGE
Les protéines n’ont pas toutes la même charge, ducoup on est obligé de les dénaturer avec du détergent, et de faire en sorte d’annuler leurs charges. Ducoup les protéines ne migrent qu’en fonction de leur taille dans un courant électrique. On peut aussi séparer les protéines par chromatographie, et séparer les fractions de protéines de tailles différentes… ce qui permet de les purifier. Et comme on sait bidouiller des protéines, on peut même leur rajouter artificiellement des bouts, et les purifier par affinité – en utilisant un principe clé/serrure. Un système souvent utilisé c’est de rajouter à une protéine un domaine dit “GST” pour gluthation S-transférase – qui reconnait spécifiquement une petite protéine, le gluthation. Ducoup, si vous mettez des billes artificielles sur lesquelles on a fixé du gluthation dans un tube, une protéine fusionnée à la GST va forcément s’accrocher aux billes, et vous pourrez la récupérer et la décrocher plus tard.
Détecter les protéines : western blot
Alors pour toutes ces méthodes de séparation sur gel, très vite les gens se sont dit – ce serait bien qu’on puisse garder les molécules sur quelque chose de plus permanent qu’un gel. Un gel ça sèche, c’est fragile, c’est chiant. Ducoup on a mis au point des techniques de transfert sur membrane. Toujours dans un champ électrique, on va fixer l’ADN, l’ARN ou les protéines sur une membrane pour toujours. On peut ensuite les révéler soit avec une “sonde” d’acide nucléique complémentaire, soit avec des trucs qui reconnaîtraient spécifiquement les protéines. Au hasard : les anticorps ! Ca tombe vraiment bien qu’on les ait découverts entre temps !
Donc les anticorps se sont des protéines immunitaires qui ont un domaine qui cible spécifiquement certains domaines d’autres protéines. Si on incube la membrane avec un anticorps dirigé contre une protéine, puis qu’on révèle ce même anticorps avec un deuxième anticorps couplé à une molécule fluorescente, ou une enzyme capable de faire une réaction colorée… on peut ducoup révéler une bande, une tâche sur la membrane qui correspond à notre protéine. grâce à ça on peut voir la présence de telle ou telle protéine dans un échantillon, sa taille… etc.
La méthode fut mise au point dans le laboratoire dans les années 80. Le nom du western blot, donné à la technique par W. Neal Burnette, est un jeu de mot à partir de la technique du Southern blot ou transfert de Southern, technique de détection d’ADN nommée d’après son inventeur, Edwin Southern et non d’après le point cardinal. La détection d’ARN est appelée northern blot ou transfert de northern. Toutes ces techniques dérivent leur nom de l’étape de transfert sur membrane, comparée à une empreinte sur buvard (blot = « tache » en anglais).
A l’époque quand j’étais passée sur le compte twitter en Direct du labo, j’avais fait un petit roman photo de la technique, concrètement en laboratoire. Je vous poste le lien dans la chatroom et sur les notes d’émission : https://wakelet.com/wake/bab38a3e-4dc3-4e56-a7ed-25f2a1e45652
Bon alors c’est génial comme méthode, et maintenant il y a plein de protéines pour lesquelles on a des anticorps. Je vous passe la méthode de production, parce que c’est pas très végan. (je prépare pour ton épisode Irène) :-) merci!
Et donc nous, biologistes moléculaires du 20 ème et 21 ème siècle, on arrive comme des enfants gâtés avec cet arsenal d’outils… mais toujours plus de questions à poser ! Puisque l’infiniment petit devient accessible, et qu’il y a une infinité de petites choses qui se passent dans cette espèce de soupe qu’est la cellule… on a une infinité de questions à poser sur comment tout fonctionne !
- Les nouvelles techniques de la biologie moléculaire
La génération des “omiques”
Un grand pan de la recherche en biologie moléculaire, c’est de déterminer quelle est la fonction d’un gène. En le clonant, en l’étudiant dans différents systèmes, in vivo et in vitro… On parle souvent de génétique moléculaire. C’est un processus lent, et on peut souvent passer une thèse à élucider le rôle de telle ou telle protéine dans tel mécanisme cellulaire ou physiologique.
Maintenant, certaines méthodes nous permettent d’analyser des changements de n’importe quoi sur l’ensemble de la cellule. Par exemple, grâce au séquençage, vous pouvez avoir la séquence d’ADN complète d’un organisme à un temps T, au nucléotide près. Grâce à la spectrométrie de masse, vous pouvez avoir le contenu en protéines total d’une cellule à un temps t. Grâce au RNA-seq vous pouvez avoir accès au contenu en ARN messagers de la cellule. Ca a l’air fou comme ça, mais tout repose sur des étapes assez simples, appliquées les unes à la suite des autres, et ça devient accessible. On est à la génération des “omiques”, des approches globales. Si vous voulez lire un article en français là dessus, je vous le poste dans la chatroom et sur les notes d’émission : http://www.ipubli.inserm.fr/bitstream/handle/10608/222/?sequence=30
Quand on étudie l’ADN global, on fait de la génomique – quand on étudie les arn messagers produits par la transcription, on fait de la transceiptomique – de la protéomique pour les protéines – et même de la métabolomique pour étudier toutes les molécules du métabolisme.
Ce qui est intéressant avec la biologie moléculaire, c’est que c’est un domaine extrêmement récent, et qui progresse sans cesse. Toutes les disciplines progressent rapidement de nos jours, évidemment, mais je trouve que c’est encore plus visible avec la biologie moléculaire.
Dans l’épisode 223 on vous parlait de la technique du CRISPR, un système d’enzymes et d’ARN bactériens qui permettent de couper virtuellement n’importe quoi. C’était y’a pas si longtemps, et maintenant on édite quasiment n’importe quel organisme, et on peut même faire des screens génomiques à grande échelle. En utilisant une librairie qui contient un ARN guide pour chacun des gènes connus, on peut créer des stratégies et identifier une liste de gènes impliquées dans un mécanisme. C’est ce que je suis entrain de faire en ce moment.
Et ducoup, je voulais faire deux trois constats pour terminer ce dossier.
- La biologie moléculaire c’est très pratico pratique. On passe notre temps avec les mains dans le cambuis. On manipule des pipettes, des tubes… et concrètement on mélange de l’eau avec de l’eau en espérant que les réactions qu’on veut se produisent.
- Avec les techniques qui évoluent, de plus en plus de choses se font par analyse bioinformatique, avec des masses de données inimaginables que seuls des gens spécialisés comme les bioinformaticiens peuvent analyser. Les progrès se font simultanément grâce au développement de nouvelles machines, de nouvelles données, et on est très transdisciplinaires.
- Mais malgré ça, les mécanismes concrets et les preuves biologiques ne peuvent se passer de l’expérimentation et ducoup… on aura encore besoin longtemps des biologistes moléculaires.
- Dernier point, même si on a des centaines de méthodes pour élucider les mécanismes de l’infiniment petit… il faut toujours se rappeler qu’on a jamais de preuve directe de ce qu’on avance. le jour où on aura des microscopes suffisamment puissants pour filmer en temps réel chaque protéine dans son environnement cellulaire, avec une étiquette sur le front – on sera certains de ce qui se passe – mais en attendant on ne peut que tester indirectement et étape par étape, construire un modèle théorique et le mettre à l’épreuve. C’est un vrai travail d’enquête au quotidien et … c’est plutôt un chouette métier.
Merci à tou.te.s d’avoir suivi ce dossier !